Arquivo Terapéutico nº 03 2018 - Hemocromatosis: o estado actual do problema

A hemocromatosis é unha patoloxía hereditaria asociada cunha alta absorción de ferro nos órganos dixestivos e a súa posterior acumulación excesiva en diversos órganos internos.
O fígado sofre máis que outros. A detección precoz da hemocromatosis, o seu diagnóstico e tratamento non permitirán o desenvolvemento de consecuencias.
Hemochromatosis - condición moderna do problema
N.B. VOLOSHINА1, M.F. OSIPENKO1, N.V. LITVINOVA1, A.N.VOLOSHIN2
1 Universidade Médica Estatal Novosibirsk FGBOU na NSMU do Ministerio de Saúde de Rusia, Rusia,
2Novosibirsk City Clinical Hospital 2, Rusia
A síndrome de sobrecarga de ferro pode asociarse a varios estados adquiridos e factores hereditarios. A hemocromatosis hereditaria é o trastorno xenético máis común. Sen intervención terapéutica, a enfermidade pode levar ao desenvolvemento de complicacións que poñen en perigo a vida como a cirrosis, o carcinoma hepatocelular. O artigo presenta datos sobre patoxénese, diagnóstico e tratamento da hemocromatosis hereditaria. Realízase observación clínica propia.
Palabras clave: hemocromatosis hereditaria, tratamento, flebotomía.
 A hemocromatosis é unha enfermidade asociada á acumulación de altos niveis patolóxicos de ferro no corpo, o que leva a trastornos funcionais dalgúns órganos. Normalmente, a absorción de ferro está fortemente regulada, como resultado do cal o corpo non é capaz de segregar exceso de ferro. O exceso de ferro acumúlase nas células como hemosiderina. Isto leva á morte das células e á substitución destas células por tecido fibroso, o que leva a interrupción da estrutura e función dos órganos. Con hemochromatosis, é posible producir danos no fígado, no páncreas, no corazón, na glándula tiroides, nas articulacións, na pel, nas gónadas e na glándula hipofisaria.
A hemocromatosis é unha enfermidade asociada á acumulación de altos niveis patolóxicos de ferro no corpo, o que leva a trastornos funcionais dalgúns órganos. Normalmente, a absorción de ferro está fortemente regulada, como resultado do cal o corpo non é capaz de segregar exceso de ferro. O exceso de ferro acumúlase nas células como hemosiderina. Isto leva á morte das células e á substitución destas células por tecido fibroso, o que leva a interrupción da estrutura e función dos órganos. Con hemochromatosis, é posible producir danos no fígado, no páncreas, no corazón, na glándula tiroides, nas articulacións, na pel, nas gónadas e na glándula hipofisaria.
A sobrecarga de ferro, que causa hemocromatosis, pode ocorrer de tres xeitos: inxestión masiva de ferro oral, aumento da absorción de ferro durante a inxestión normal de ferro e produción excesiva ou masiva transfusión de glóbulos vermellos.
Na hemocromatosis hereditaria, o exceso de ferro deposítase normalmente nas células parenquimáticas, mentres que na hemocromatosis transfusional deposítase principalmente nas células reticuloendoteliais 1-3.
A hemocromatosis hereditaria inclúe un grupo de trastornos xenéticos caracterizados por unha maior absorción de ferro. O mecanismo predominante na maioría dos tipos de hemocromatosis hereditaria é o efecto hepcidina, que desempeña un papel clave na homeostase de ferro 4-6. A hepsidina sintetízase principalmente en hepatocitos e controla a concentración de ferro no plasma ao unirse a ferroportina (tamén chamada SLC40A1), o único transportista transmembrana de ferro procedente de tecidos do donante de ferro. A ferroportina exporta ferro do duodeno, dos macrófagos e dos hepatocitos.
No plasma, o ferro únese á transferrina, polo que a saturación de ferro coa transferrina é de media do 35% (valor medio da mañá). A hepsidina inhibe a liberación de ferro dos macrófagos (de vellos glóbulos vermellos e ferritina), hepatocitos e enterocitos duodenais uníndose a ferroportina. E a falta de ferroportina, a saída de ferro de enterocitos, hepatocitos e macrófagos está bloqueada. Así, a hepcidina reduce a absorción de ferro no intestino, reduce o nivel de ferro liberado de hepatocitos e macrófagos, o que leva a un baixo nivel de ferro no plasma e un aumento dos tecidos.
A causa da hemocromatosis hereditaria é unha mutación no xene HFE. O defecto do xene HFE describiuse por primeira vez en 1996, o que supón unha mutación que conduce á substitución da tirosina por cisteína na posición 282 do aminoácido (C282Y). Unha mutación no xene HFE provoca unha maior absorción de ferro, a pesar da inxestión normal de ferro. A proteína HFE regula a produción de hepcidina. Os pacientes con homocigotos C282Y hemochromatosis hereditarios son do 80 ao 85% 1, 8.
Hai dúas mutacións máis: unha está asociada coa substitución de aspartato por histidina na posición 63 (H63D), e a segunda é unha substitución da cisteína coa serina na posición 65 (S65C). Estas mutacións non están relacionadas coa síndrome de sobrecarga de ferro, a non ser que C282Y forme parte integrante dos heterozigos C282Y / H63D ou C282Y / S65C. Así, a forma de hemocromatosis hereditaria asociada ao HFE pode comprobarse cun curso asintomático da enfermidade. Por conseguinte, pódese aplicar un diagnóstico xenético en pacientes nos que a hemocromatosis aínda non se manifestou fenotípicamente. Este grupo de pacientes con predisposición xenética á hemocromatosis. Os heterocigotos teñen un maior risco de desenvolver diabete en comparación coa poboación xeral, descoñecendo o mecanismo de desenvolvemento 9-11.
Previamente pensábase que en todos os pacientes cun defecto xénico HFE, co tempo se desenvolverá unha clínica de hemocromatosis. Non obstante, agora comprobouse que a expresión fenotípica só se atopa en aproximadamente o 70% dos homocigotos C282Y, e menos do 10% deles desenvolven unha sobrecarga de ferro grave con danos nos órganos internos 12, 13.
A táboa mostra a clasificación das síndromes de sobrecarga de ferro segundo a causa da súa aparición.
Dependendo da causa da enfermidade, os pacientes con síndrome de sobrecarga de ferro pódense dividir en 4 grupos: pacientes con hemocromatosis hereditaria, pacientes con hemocromatosis secundaria provocados por varias causas e un pequeno grupo de pacientes, que destaca como "diferente".
A causa da hemocromatosis secundaria é a hemocromatosis eritropoietica. Na maioría das veces isto ocorre como resultado dunha enfermidade sanguínea subxacente na que os glóbulos vermellos teñen unha vida útil máis curta. Este grupo de enfermidades inclúe anemia con deficiencia de ferro, talasemia, anemia sideroblástica, anemia hemolítica crónica, anemia aplástica, anemia sensible á piridoxina, deficiencia de piruvato quinasa.
A síndrome de sobrecarga de ferro pode ocorrer en pacientes que reciben transfusións prolongadas e múltiples de glóbulos vermellos. Como se pode ver na táboa, outras enfermidades bastante raras, como por exemplo a porfiria, tamén poden causar a síndrome de sobrecarga de ferro.
Finalmente, a inxestión excesiva de ferro pode causar hemocromatosis. Feito histórico coñecido: o uso da cervexa feita en tambores de aceiro foi a causa da síndrome de sobrecarga de ferro. Ademais, unha sobredose de preparados de ferro pode causar a síndrome de sobrecarga de ferro. Hai que lembrar que moitos suplementos nutricionais sen receita conteñen ferro nunha dosificación suficientemente grande, polo que o seu uso incontrolado é inaceptable.
Os síntomas da enfermidade dependen do órgano máis afectado. Non obstante, case todos os pacientes quéixanse de fatiga e fatiga importantes. Non hai síntomas específicos de hemocromatosis. A maioría das veces, o diagnóstico faise na fase da enfermidade, cando varios sistemas xa foron afectados. Desde os primeiros síntomas da enfermidade ata a verificación do diagnóstico normalmente leva polo menos dez anos. En mulleres con hemocromatosis, os síntomas da enfermidade maniféstanse a unha idade máis recente que nos homes, debido á perda de sangue menstrual, a perda de “ferro materno” durante o embarazo e o efecto antioxidante do estróxeno, e a enfermidade non se manifesta clínicamente antes do período climático.
Aproximadamente o 50% dos pacientes con síntomas de hemocromatosis hereditaria teñen diabetes mellitus, o risco de que se produza aumente significativamente nos heterozigotos. A cirrosis hepática está presente no 70% dos pacientes con hemocromatosis. Neste grupo de pacientes, aumenta notablemente a incidencia de carcinoma hepatocelular, que é a principal causa de morte.
O dano nas articulacións coa hemochromatosis maniféstase en forma de artralxia (normalmente a segunda e a terceira articulacións metacarpopánxicas). Normalmente non se producen deformidades articulares coa hecocromatosis, aínda que son posibles cambios de dexeneración nas articulacións. Nestes pacientes, por regra xeral, pódense atopar cristais de pirofosfato de calcio no fluído sinovial. É característico da poliartrite con hemocromatosis que incluso despois de normalizar as tendas de ferro aínda pode progresar.
A deposición de ferro nas fibras do músculo cardíaco e as células do sistema de condución do corazón pode provocar alteracións do ritmo cardíaco e / ou cardiomiopatía dilatada, con máis desenvolvemento de insuficiencia cardíaca. Nalgúns casos, hai unha compensación completa pola falla ventricular esquerda despois de normalizar o nivel de ferro no corpo 9-12.
Con hemochromatosis, é posible o desenvolvemento de hipogonadismo e, en consecuencia, a impotencia por insuficiencia hipotalámica e / ou hipofisaria, que orixina unha violación da liberación da hormona gonadotropina. En casos de exceso de ferro almacenado cinco veces ou máis, prodúcese hiperpigmentación da pel, o que é o resultado da deposición de ferro e melanina. A sobrecarga de ferro dos macrófagos pode provocar unha fagocitose deteriorada e unha diminución da inmunidade, o que leva a un maior risco de infección por Listeria, Yersinia enterocolitica e Vibrio vulnificus. A deposición de ferro na glándula tiroides normalmente provoca hipotiroidismo.
A etapa desenvolvida da hemocromatosis caracterízase pola presenza de cirrosis, diabetes mellitus e pigmentación da pel (a chamada diabetes de bronce). En pacientes que abusan do alcol e están infectados con hepatite B e / ou C, a patoloxía do fígado e do páncreas asociada á hemocromatosis procede significativamente dun xeito máis grave 1-3.
O diagrama mostra medidas diagnósticas para sospeita de hemocromatosis. Sábese que só preto do 70% dos homocigotos C282Y teñen niveis elevados de ferritina, o que corresponde a un aumento das tendas de ferro, e só unha pequena porcentaxe destes pacientes presentan manifestacións clínicas da enfermidade. Por suposto, todos os pacientes con síntomas que poidan ocorrer con hemocromatosis deben someterse a un exame adicional para excluír a enfermidade. Débese prestar especial atención aos pacientes con debilidade non motivada, artralxia, dor no cuadrante superior dereito do abdome, impotencia, diminución da libido, síndrome de insuficiencia cardíaca, pigmentación da pel e diabetes. Ademais, en todos os pacientes con hepatomegalía, a síndrome citolítica, coa etapa cirrosa da enfermidade, é necesario, ademais de todas as posibles causas etiolóxicas da enfermidade, lembrar a posibilidade de hemocromatosis. Por suposto, a hemocromatosis hereditaria debería excluírse en pacientes con parentes do primeiro grao de parentesco que padecen hemochromatosis. 
O estudo debería comezar medindo a saturación da transferrina sérica ou da concentración sérica de ferritina. Cómpre sinalar que a determinación da transferrina nos casos de hemocromatosis eritropoetica non é tan eficaz para a comprobación da síndrome de sobrecarga de ferro. A especificidade da ferritina depende en gran medida da presenza de enfermidades inflamatorias. Se o nivel de ferritina é superior a 200 μg / l en mulleres ou 300 μg / l en homes ou a saturación de transferrina supera o 40% en mulleres ou o 50% en homes, é necesario facer máis probas para excluír a hemocromatosis 1,2, 10, 11.
Segundo as recomendacións da Asociación Americana para o Estudo das Enfermidades do Fígado 2011 (AASLD 2011) se o paciente ten unha transferrina sérica de 1000 mg / l), e segundo estes indicadores, tómase unha decisión sobre a táctica terapéutica e a necesidade dunha biopsia hepática. )
En pacientes con combinación de heterocigotos C288Y / H63D, así como heterocigotos C288Y ou non C288Y, é necesaria unha eliminación minuciosa doutras enfermidades do fígado ou do sangue (se é necesario, é necesaria unha biopsia de punción do fígado) e, a continuación, faise unha sangría médica.
Non hai probas fiables de que determinadas dietas afecten ao inicio ou á progresión da hemocromatosis. Non obstante, algúns autores cren que aos pacientes con hemocromatosis hereditaria móstrase unha dieta a excepción do té e cítricos, que, na súa opinión, contribúen á acumulación de ferro. Por suposto, o alcohol, que é a principal sustancia hepatotóxica, debería estar estrictamente prohibido para os pacientes con hemocromatosis. Ademais, o etanol demostrou que reduce a síntese de hepcidina 20, 21.
O tratamento principal para a hemocromatosis primaria é o líquido sanguíneo. Reducir o número de glóbulos vermellos, que son o principal mobilizador do ferro no corpo, reducindo e minimizando o efecto tóxico do ferro. Os pacientes poden requirir 50 a 100 sangreis ao ano, 500 ml cada un, para baixar os niveis de ferro á normalidade. Unha vez que o nivel de ferro está normalizado, é necesaria unha limpeza de toda a vida, pero menos frecuente, normalmente 3-4 veces ao ano. O obxectivo do líquido sanguíneo é manter os niveis de ferritina de 50 a 100 µg / L. En casos de diminución significativa da hemoglobina despois do líquido sanguíneo, é recomendable o tratamento conxunto con eritropoyetina.
Se se detecta unha hemocromatosis nun estadio temperán da enfermidade, o tratamento sanguíneo pode evitar a disfunción dos órganos afectados e aumentar así a esperanza de vida do paciente. Non obstante, os pacientes raramente viven máis de dous anos despois do diagnóstico, nos casos de diagnóstico tardío no estadio de manifestacións clínicas detalladas 22, 23.
Segundo a Asociación Europea para o Estudo do Fígado (EASL 2010), as indicacións para a sangría terapéutica son niveis elevados de ferritina sérica. Recoméndase que o líquido sanguíneo terapéutico cun volume de 400-500 ml se realice unha vez á semana ou unha vez cada 2 semanas ata que se alcance un nivel de ferritina do 45% e un aumento significativo da ferritina sérica ata 1444 mcg / l, o diagnóstico de hemocromatosis é innegable. Analizáronse mostras de ADN para mutacións no xene HFE - atopouse unha mutación C282Y (c.845 G> A) no estado homocigoto s.845A / s.845 A.
Así, o diagnóstico do paciente K. é hemocromatosis hereditaria, unha mutación homozigota no xene HFE (C288Y / C288Y) con dano hepático predominante, fibrose de grao 1 (FibroScan, Metavir 6,6 kPa).
A manifestación tardía e o diagnóstico da enfermidade aos 58 anos de idade en 2015 débese á compensación a longo prazo da enfermidade debido á perda de sangue masiva debido ao sangue menstrual, doazón de sangue e perda de sangue durante a terminación do embarazo e do parto.
Cabe destacar que pasaron 8 anos dende a aparición dos primeiros signos da enfermidade á verificación do diagnóstico. Desde finais de 2015, ao paciente se lle prescribiu terapia: líquido sanguíneo de 500 ml unha vez por semana. O paciente toleraba ben o líquido sanguíneo, observou unha mellora significativa das condicións despois do primeiro procedemento. Controlouse un exame xeral de sangue e ferritina sanguínea, cuxo nivel diminuíu gradualmente. En total, máis de 100 líquidos sanguíneos realizáronse en 2 anos. Non obstante, ata o momento non se logrou o nivel de transferrina obxectivo (100 μg / l) debido a que o paciente salta periodicamente o procedemento, explicándolle a boa saúde. Na actualidade, o paciente continúa a terapia, logrou convencelo da necesidade dunha terapia ao longo da vida.
Así, hai que recordar que en presenza de síndrome citolítico en pacientes, a investigación diagnóstica debe incluírse na hemocromatosis hereditaria. A terapia de elección para a hemocromatosis hereditaria segue sendo sanguenta. A terapia adecuada iniciada no tempo permite evitar o desenvolvemento da etapa cirrótica da enfermidade e aumentar así a esperanza de vida dos pacientes.
Información sobre os autores:
Voloshina Natalya Borisovna - candidato de ciencias médicas, profesor asociado propedéuticos das enfermidades internas da facultade médica
Osipenko Marina Fedorovna - doutor en ciencias médicas, prof., xefe. cafetería propedéuticos das enfermidades internas da facultade médica
Voloshin Andrey Nikolaevich - Médico do Hospital Clínico Novosibirsk City 2
Hocromromatosis: que é esta enfermidade?
Para comprender a esencia da enfermidade, debes saber canto ferro debería ter normalmente unha persoa. Nos homes, o ferro é de aproximadamente 500-1500 mg, e nas mulleres, de 300 a 1000 mg. Os indicadores non só dependen do xénero, senón tamén do peso da persoa. Máis da metade da cantidade total de ferro está na hemoglobina.
Ao redor de 20 mg deste microelemento entra no corpo con alimentos ao día. Destes, só 1-1,5 mg é absorbido no intestino. Con hemocromatosis (GC) ou siderofilia, como tamén se denomina esta enfermidade, a absorción aumenta ata os 4 mg diarios e o ferro acumúlase gradualmente nos tecidos de varios órganos.
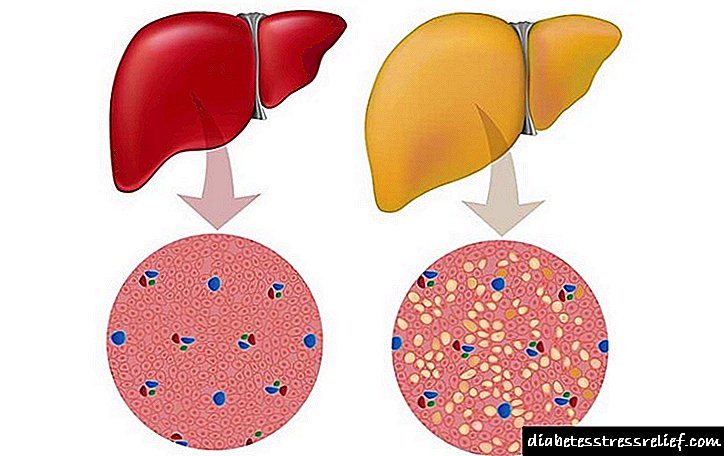
Fígado saudable e hemocromatosis
O seu exceso destrúe as moléculas de proteínas e carbohidratos, e de aí o propio órgano. En pacientes con GC, a cantidade de ferro no fígado pode alcanzar o 1% da masa seca do órgano, que está chea de cirrose, e nun terzo dos casos con cancro de fígado. Danado polo exceso de ferro, o páncreas pode dar un impulso ao desenvolvemento da diabetes.
Ao depositarse na glándula pituitaria, o ferro destrúe todo o sistema endócrino. Os órganos reprodutivos sofren máis que outros: os homes teñen disfunción eréctil e as mulleres poden desenvolver infertilidade.
Causas
A principal razón para o GC é o "mal funcionamento" do xene, ou mellor dito, do xen HFE. É el quen regula o curso dos procesos químicos e a cantidade de ferro que entra no corpo como parte dos alimentos. A mutación que se produce nel conduce á interrupción do metabolismo do ferro.
Outras causas de GC son:
- talasemia. Neste caso, a estrutura da hemoglobina destrúese coa liberación de ferro,
- hepatite
- o ferro pode aumentar como consecuencia das transfusións de sangue frecuentes. O feito é que a vida útil dos glóbulos vermellos alieníxenas é moito máis curta que a propia. Cando morren, liberan ferro,
- procedementos de hemodiálise.
Clasificación e código ICD-10
No clasificador xeralmente aceptado de enfermidades de GC, atribúeselle o código E83.1.
Nunha liña etilóxica, distínguense primaria (ou GC hereditaria) e secundaria:
- primario. Este tipo de enfermidade ten un carácter hereditario e é o resultado dun defecto no sistema encimático que afecta ao metabolismo do ferro. Está diagnosticado en 3 persoas de cada 1000. Obsérvase que os homes son máis susceptibles a esta patoloxía e padecen dela 3 veces máis que as mulleres,
- secundario. A súa causa son as enfermidades hepáticas do paciente (que adoita observarse con alcoholismo), a transfusión de sangue, o autotratamento con complexos vitamínicos cun alto contido en ferro. A causa do GC adquirido pode ser problemas de pel e enfermidades do sangue.
A hemocromatosis primaria (PCH) caracterízase por un desenvolvemento gradual e, nos primeiros estadios, os pacientes quéixanse de fatiga. Pódense molestar pola dor no lado dereito e a pel seca.
A etapa ampliada de PCH caracterízase por:
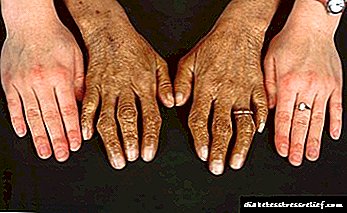
- pigmentación específica da cara, pescozo, brazos e axilas. Adquiren unha tonalidade de bronce,
- cirrosis do fígado. Está diagnosticado nun 95% dos casos,
- insuficiencia cardíaca
- artrite
- diabetes mellitus: nun 50% dos casos,
- bazo engrandido,
- disfunción sexual.
Nas últimas etapas obsérvase hipertensión portal e ascite. Pode producirse cancro de fígado.
 Dado que o exceso de ferro fórmase co paso dos anos, os síntomas iniciais do GC secundario maniféstanse nos homes despois dos 40 anos e nas mulleres despois dos 60 anos.
Dado que o exceso de ferro fórmase co paso dos anos, os síntomas iniciais do GC secundario maniféstanse nos homes despois dos 40 anos e nas mulleres despois dos 60 anos.
Os síntomas son os seguintes:
- melasma,
- fatiga e perda de peso,
- diminución da libido
- ampliación e densificación do tecido hepático,
- cirrosis (na última etapa do GC).
Proba de sangue e outros métodos de diagnóstico
 Un gastroenterólogo confirma o diagnóstico. Nas primeiras etapas da enfermidade, as probas de laboratorio son moi importantes.
Un gastroenterólogo confirma o diagnóstico. Nas primeiras etapas da enfermidade, as probas de laboratorio son moi importantes.
Con GC, realízanse análises de sangue especiais para detectar os valores do ferro no plasma, a súa baixa capacidade de unión ao ferro e a saturación coa transferrina.
O principal síntoma da enfermidade son os depósitos de hemosiderina nos hepatocitos do fígado, na pel e noutros órganos, que quedan "enferruxados" debido ao exceso deste pigmento. Tamén se precisa unha proba de sangue xeral para a bioquímica, así como o azucre. Ademais, tómanse probas hepáticas.
 Ademais, tamén se realizan estudos instrumentais:
Ademais, tamén se realizan estudos instrumentais:
- A biopsia do fígado é o principal xeito de confirmar o GC,
- Ecografía do abdome
- Resonancia magnética do fígado (nalgúns casos)
- ecocardiografía, para excluír / confirmar cardiomiopatía,
- radiografía conxunta.
Dieta terapéutica
É importante entender que cunha hemocromatosis diagnosticada, a dieta debe ser toda a vida.
A regra principal é a redución máxima na dieta de produtos que conteñen ferro, especialmente:

- queixos duros e peixes de mar,
- cereais: avea, millo e trigo mouro,
- pan negro
- legumes e froitos secos,
- ácido ascórbico e drogas cun alto contido en vitamina C,
- os excrementos, especialmente o fígado, están completamente excluídos.
O alcol é un tabú absoluto. Pero o té e o café móstranse ao contrario. Teñen tanino, que ralentiza a absorción de ferro.
Lista de medicamentos utilizados
Este tratamento realízase con medicamentos que eliminan o ferro do corpo do paciente. Na fase inicial prescríbense vitaminas A, E e ácido fólico. A continuación úsanse quelantes (como Desferal).

Dosificación por inxección: 1g / día. Xa 500 mg de fármaco dan un resultado tanxible: se excretan ata 43 mg de ferro. O curso ten unha duración de ata 1,5 meses. O uso prolongado é perigoso: é posible que se poida anubrar as lentes.
Phlebotomía e outros procedementos terapéuticos
 A flebotomía é o tratamento non farmacolóxico máis sinxelo e, ao mesmo tempo, bastante eficaz da GC.
A flebotomía é o tratamento non farmacolóxico máis sinxelo e, ao mesmo tempo, bastante eficaz da GC.
Unha punción faise na vea do paciente e a sangue libérase do corpo. Drenan uns 500 ml por semana.
O procedemento é só ambulatorio. A sangue é constantemente probado para a concentración de ferrina: debe baixar a 50. Isto pode levar 2-3 anos. Ademais, a terapia está dirixida a manter o valor óptimo deste oligoelemento.
Tratamento con remedios populares
Esta terapia ten un efecto leve sobre os órganos enfermos.
Tratamento do fígado:

- cabaza. É bo tanto cru coma cocido. Os vexetais engádense ás ensaladas ou mestúranse con mel - saborosos e saudables! Tamén se mostra o zume de cabaza: medio vaso cun estómago baleiro,
- remolacha- Outro produto útil para GC. Use en forma crúa ou fervida. Zume saudable e recén espremido.
Para o tratamento cardíaco, pode aconsellar infusións de espino, adonis ou patria nai. As herbas vertéronse con auga fervendo e, despois de insistir, bótanse segundo as instrucións.
Tratamento do páncreas:

- A decocción de sementes de plátano axudará. Proporcións: 1 cda. materias primas a 1 cda. auga. As sementes elaboradas son fervidas durante 5 minutos, arrefriadas e tomadas antes das comidas, 1 cda.,
- mel con canela. Proporcións: 1 cda. en po a 1 colher de sopa de auga. Insista 15-30 minutos. e engade un pouco de mel. Deixar outras 2 horas. Todos os medios deben ser bebidos nun día.
Harina de avena útil e non cociñada (con casca). Proporcións: 100 g de cereal a 1,5 litros de auga. Ferva durante polo menos media hora. A continuación, xusto no recipiente onde se cociñou a avea, esmagámola ata que se esmoreza e volva ferver durante 40 minutos. A vida do caldo filtrado non será superior a 2 días. Beba medio vaso antes das comidas.
Prognóstico e principais pautas clínicas
Pero se a terapia realízase baixo supervisión médica e puntualmente, a vida do paciente aumenta significativamente.
Sendo unha enfermidade hereditaria, a hemocromatosis no 25% dos casos diagnostícase nos parentes do paciente. Así, deben examinarse máis. Isto revelará a enfermidade incluso antes das manifestacións clínicas e no futuro evitará as súas complicacións.
No caso de GC secundario, a dieta é recomendable, é importante manter o estado do fígado e do sangue baixo control. A hemocromatosis detectada durante o embarazo (ou na fase de planificación) non é perigosa.
Vídeos relacionados
Sobre os síntomas, causas e métodos de tratamento da hemocromatosis no vídeo:
Por desgraza, aínda non se identificou a causa raíz da hemocromatosis. Pero na actualidade elaborouse e utilízase activamente unha técnica especial de tratamento integral, co obxectivo de interromper as manifestacións clínicas da enfermidade e reducir o risco das súas posibles complicacións.
- Estabiliza os niveis de azucre durante moito tempo
- Restablece a produción de insulina pancreática
Coñece máis. Non é unha droga. ->
Terapia con enfermidades concomitantes
O exceso de ferro nos órganos leva ao desenvolvemento de múltiples patoloxías. Todos eles precisan terapia adxuvante. Por exemplo, se o GC contribuíu ao desenvolvemento da diabetes, este último debe ser tratado, sempre controlando a taxa de azucre.
Se se detectan patoloxías no fígado, o seu tratamento continúa. Isto é necesario para evitar o desenvolvemento da patoloxía ao estado dun tumor maligno.
Hemocromatosis
A hemocromatosis hereditaria (NG) é unha enfermidade polisistémica baseada en trastornos metabólicos do ferro determinados xeneticamente, o que leva á súa acumulación excesiva no corpo e danos tóxicos a órganos e tecidos.
A primeira descrición da enfermidade pertence a A. Trousseau (1865), que identificou unha tríada das principais manifestacións clínicas: diabetes mellitus, pigmentación na pel de bronce, cirrosis. O termo "hemocromatosis" foi proposto en 1889 pola F.D. von Recklinghausen. Desde 1935, a enfermidade pertence ao grupo de enfermidades hereditarias. En 1996, J.N. Feder et al. identificou o xene para hemocromatosis hereditaria (HFE), mutacións das cales a maioría das veces conducen ao desenvolvemento desta enfermidade. En 2000-2004 Descríbense mutacións doutros xenes que conducen ao desenvolvemento da hemocromatosis.
A prevalencia da enfermidade varía entre 1: 250 individuos que viven no norte de Europa a 1: 3300 entre a poboación negra dos Estados Unidos e os países africanos. A enfermidade é diagnosticada en homes 5-10 veces máis veces que nas mulleres. Durante o cribado xenético, descubriuse que unha mutación homozigota do xen HFE se detecta en 1 de cada 500 pacientes examinados, mentres que o número de casos clínicos de NG establecidos é de 1: 5.000. Así, un número significativo de casos da enfermidade non se recoñecen nin se diagnostican tarde, na fase de danos internos irreversibles. órganos (cirrosis, diabetes mellitus, cardiomiopatía dilatada).
De acordo coa base xenética da enfermidade, distínguense 4 tipos de hemocromatosis hereditaria:
Tipo I - herdado por un mecanismo autosómico recesivo, debido a mutacións no xene HFE situado no cromosoma 6. Na maioría das veces (no 87-90% dos pacientes), rexístrase a mutación C282Y: a substitución da cisteína pola tirosina no 282º aminoácido. A mutación H63D é menos común: a substitución de citidina por guanina no 63 aminoácido,
O tipo II - a hemocromatosis xuvenil é rara, debido a mutacións no xene responsable da síntese doutra proteína do metabolismo do ferro - hepsidina,
Tipo III: a base xenética consiste en mutacións dun xene que codifica a síntese do receptor da transferrina,
Tipo IV: a base xenética consiste en mutacións no xene SLC40A1, que codifica a síntese da proteína de transporte ferroportina.
Etioloxía e patoxénese
O ferro é un compoñente bioquímico necesario dos procesos metabólicos máis importantes, por unha banda, e é un elemento potencialmente tóxico que pode causar danos oxidativos ás membranas biolóxicas, ás proteínas e aos ácidos nucleicos, por outro. De acordo con isto, a homeostase do ferro no corpo humano está fortemente regulada. A maior parte deste elemento sofre un proceso de reciclaxe: os macrófagos do bazo e o fígado captan e destruen os glóbulos vermellos envellecidos, degradan a hemoglobina e liberan o ferro, que se une á transferrina ou á ferritina e reciclase. A perda fisiolóxica diaria de ferro non supera os 1-2 mg e compénsase coa absorción dunha cantidade equivalente de ferro no tracto gastrointestinal. Non hai mecanismos que controlen a eliminación de ferro nos humanos.
As mutacións dos xenes responsables da síntese de proteínas implicadas no metabolismo do ferro provocan un desequilibrio entre a inxestión e a perda de ferro, a acumulación patolóxica deste elemento en órganos e tecidos e a aparición de ferro libre (non asociado coa transferrina) no sangue. O desenvolvemento da hemocromatosis tipo I está asociado a unha mutación do xene responsable da síntese da proteína HFE (proteína de hemocromatosis), que é unha glicoproteína (MM = 37.235 daltons), similar en estrutura ás proteínas do principal complexo de histocompatibilidade da clase 1. A función da proteína HFE no metabolismo do ferro e o mecanismo dun forte aumento da absorción de ferro durante as mutacións no xene HFE non foron completamente establecidos.
A patoxénese da hemocromatosis tipo II-IV está asociada a mutacións en xenes que codifican outras proteínas implicadas no metabolismo do ferro - hepsidina, receptor da transferrina-II, ferroportina.
Unha característica distintiva do tipo IV NG, que está baseada en mutacións do xene ferroportina, é unha violación predominante dos procesos de recirculación de ferro, que fenotípicamente se manifesta como anemia hipocrómica profunda e eritropoiese con deficiencia de ferro en combinación con hemocromatosis severa de órganos internos.
A acumulación patolóxica de ferro en órganos parénquima está asociada a cambios dexenerativos do parénquima celular e ao desenvolvemento progresivo do tecido fibroso, o que leva a unha disfunción irreversible dos órganos vitais. Os órganos diana máis vulnerables son o fígado, o corazón e o páncreas.
Sinais e síntomas clínicos
O cadro clínico de NG está determinado polo nivel de acumulación de ferro en órganos e tecidos. Con hipertensión tipo I, as manifestacións clínicas normalmente atópanse aos 45-50 anos e máis. Na hemocromatosis xuvenil (tipo II), as lesións graves no fígado e no corazón aparecen cedo - na segunda ou terceira década da vida. Nos homes, as manifestacións clínicas da enfermidade obsérvanse 3 veces máis veces que nas mulleres, o que está asociado ás características fisiolóxicas do corpo feminino. As principais manifestacións clínicas inclúen síntomas de dano ao fígado, corazón, órganos do sistema endócrino e articulacións.
 Pódense detectar signos de dano hepático durante un exame aleatorio en forma de aumento desmotivado de transaminases ou debut con síntomas de hipertensión portal: ascite, hepatosplenomegalia, sangrado por varices do esófago e estómago.
Pódense detectar signos de dano hepático durante un exame aleatorio en forma de aumento desmotivado de transaminases ou debut con síntomas de hipertensión portal: ascite, hepatosplenomegalia, sangrado por varices do esófago e estómago.
Os síntomas do dano cardíaco inclúen ataques cardíacos, desenvolvemento de arritmias e signos de insuficiencia cardíaca. A cardiomiopatía grave é a principal causa de morte en pacientes novos.
O desenvolvemento da diabetes e a disfunción das glándulas xenitais son síntomas característicos da NG. Nos homes, atrofia testicular, diminución da pulsión sexual, impotencia, azoospermia son frecuentemente observadas, nas mulleres - amenorrea, infertilidade.
O dano nas articulacións maniféstase por artralxia persistente, as articulacións metacarpofalángeas están máis a miúdo implicadas, menos frecuentemente as articulacións do xeonllo, da cadeira e do cóbado. A rigidez das articulacións desenvólvese gradualmente.
Outras manifestacións clínicas de NG inclúen unha marcada debilidade desmotivada, fatiga, somnolencia, ataques de dor abdominal de diversa intensidade e localización, hiperpigmentación cutánea e tendencia a varias infeccións (incluíndo microorganismos que raramente afectan a persoas saudables - Yersenia enterocolitica e Vibrio vulnificus).
O diagnóstico de NG establécese a partir dun cadro clínico e de laboratorio característico.É fácil sospeitar do diagnóstico de hemocromatosis nun paciente cunha combinación dos seguintes síntomas: artralxia, dor abdominal, pel gris bronceada, presenza de diabetes mellitus e hepatomegalia.
Exame de sangue: é característica unha combinación dun nivel alto de hemoglobina cunha baixa concentración de hemoglobina en eritrocitos (MCH). O desenvolvemento de anemia ou outra citopenia obsérvase nas etapas tardías da enfermidade - en pacientes con cirrosis do fígado ou é o resultado de numerosas hemorraxias.
Estudo do metabolismo do ferro necesario para identificar os signos de laboratorio de sobrecarga de ferro e inclúe a determinación do nivel de ferro, ferritina e transferrina do soro sanguíneo, a capacidade total de unión do ferro do soro (OZHSS) e o coeficiente de saturación de transferrina estimada do ferro (NTZH). A NG caracterízase por un aumento dos niveis de ferro e de ferritina sérica, unha diminución dos niveis de OGSS e transferrina. Un signo importante de hemocromatosis é un aumento do coeficiente de ITS en homes por riba do 60%, en mulleres - por riba do 50%.
Proba desferal confirma a presenza de sobrecarga de ferro: despois de 0,5 g intramusculares de deferoxamina (desferal), a excreción diaria de ferro en orina supera significativamente o nivel normal (0-5 mmol / día).
No tipo NG IV, o cadro de laboratorio pode ser representado por anemia hipocrómica profunda, hiposiderinemia e elevada ferritina sérica, que se combina coa sobrecarga de tecidos severa con ferro.
Realización de análises xenéticas moleculares permite confirmar a natureza hereditaria da hemocromatosis e excluír a natureza secundaria da sobrecarga de ferro. O diagnóstico de NG establécese en presenza de mutacións homocigotas do xene HFE (C282Y ou H63D) ou cando se detectan heterocigotos complexos (unha combinación de mutacións heterozigotas C282Y e H63D) en pacientes con signos de laboratorio de sobrecarga de ferro. As mutacións heterocigotas illadas C282Y e H63D atópanse na poboación de persoas saudables cunha frecuencia do 10,6% e o 23,4% dos casos, respectivamente, a presenza destas mutacións non é a base para o diagnóstico de NG.
Toma de TC de órganos abdominais revela unha maior densidade de tecido hepático debido aos depósitos de ferro e permite sospeitar a presenza de hemocromatosis.
Con resonancia magnética o fígado dun paciente con hemocromatosis ten unha cor gris escura ou negra. A TC e a resonancia magnética do fígado son necesarias para excluír o diagnóstico do carcinoma hepatocelular.
Biopsia hepática cunha determinación semicantitativa ou cuantitativa do contido de ferro permítelle determinar o grao de desenvolvemento da fibrose e a concentración de ferro no tecido hepático. Para o diagnóstico de hemocromatosis, recoméndase calcular o "índice hepático de ferro", que é igual á relación do contido de ferro no tecido hepático (en micromol / g de peso seco) coa idade do paciente (en anos). Un índice> 2.0 confirma o diagnóstico de NG.
A hemocromatosis hereditaria debe diferenciarse con síndromes secundarias de sobrecarga de ferro, que se desenvolven en pacientes con anemia hemolítica hereditaria e adquirida, algunhas formas de síndrome mielodisplásica (anemia sideroblástica refractaria), porfiria, así como en pacientes con dano alcohólico no fígado.
O obxectivo do tratamento da NG é eliminar o exceso de ferro do corpo e evitar danos irreversibles nos órganos internos. Un método común de tratamento é o líquido sanguíneo. O curso inicial consiste en líquido sanguíneo nun volume de 500 ml unha vez por semana. Despois de baixar o nivel de hemoglobina en 15-20 g / l, o nivel de MCV en 3-5 fl. e o contido de ferritina sérica de ata 20-50 ng / ml, vai á terapia de mantemento: eliminación de 500 ml de sangue cada 2-4 meses nos homes e cada 3-6 meses nas mulleres. O tratamento é permanente.
En presenza de anemia ou outras contra-indicacións (por exemplo, insuficiencia cardíaca), os quelantes de ferro úsanse para a limpeza de sangue. A deferoxamina une o exceso de ferro nos tecidos e o soro sanguíneo e excreta coa orina e as feces. Non obstante, a vida media deste medicamento é curta - só 10 minutos, o que require unha administración lenta: intravenosamente en forma de infusións de 3-4 horas ou de forma subcutánea, preferiblemente en forma de infusións de 12 horas ou as 24 horas con bombas especiais. Desenvolvéronse novos fármacos formadores de complexos para a administración oral e están na fase de estudo ou implementación clínica, dos que o máis eficaz é o Deferasirox.
A eficacia do tratamento está determinada pola dinámica de datos clínicos e de laboratorio. A condición dos pacientes comeza a mellorar despois dun curso de hemorragia: a debilidade, a fatiga, a somnolencia desaparecen, o tamaño do fígado diminúe, o curso da diabetes e a cardiomiopatía poden mellorar. O control de laboratorio inclúe o estudo de hemograma, indicadores de ferritina, ferro e NTZH (1 vez en 3 meses), o nivel de excreción urinaria de ferro.
No caso dun diagnóstico precoz de hipertensión e unha limpeza de sangue terapéutica oportuna, o prognóstico é favorable: a esperanza de vida dos pacientes non difire da esperanza de vida das persoas que non padecen hemochromatosis. En casos de diagnóstico tardío da enfermidade, en presenza de cirrosis hepática, cardiomiopatía, diabetes mellitus, o prognóstico está determinado pola gravidade destas complicacións irreversibles. As principais causas de morte dos pacientes son: complicacións de diabetes, insuficiencia cardíaca, cancro de fígado primario, insuficiencia hepática, hemorraxias por varices do esófago e estómago, infeccións intercurrentes.
Información xeral
A hemocromatosis (diabetes de bronce, cirrosis pigmentaria) é unha violación xeneticamente causada polo metabolismo do ferro, provocando a deposición de pigmentos que conteñen ferro en tecidos e órganos e o desenvolvemento dunha falla de órganos múltiples. A enfermidade, acompañada dun complexo síntoma característico (pigmentación da pel, cirrosis hepática e diabetes mellitus) foi descrita en 1871, e en 1889 chamouse hemocromatosis pola cor característica da pel e dos órganos internos. A frecuencia da hemocromatosis hereditaria nunha poboación é de 1,5-3 casos por cada 1000 habitantes. Os homes sofren de hemocromatosis 2-3 veces máis veces que as mulleres. A idade media do desenvolvemento da patoloxía é de 40-60 anos. Debido á natureza polisistémica da lesión, diversas disciplinas clínicas están implicadas no estudo da hemocromatosis: gastroenteroloxía, cardioloxía, endocrinoloxía, reumatoloxía, etc.
No aspecto etiolóxico distínguense hemocromatosis primaria (hereditaria) e secundaria. A hemocromatosis primaria está asociada a un defecto nos sistemas encimáticos, o que conduce á deposición de ferro nos órganos internos. Dependendo do defecto xénico e do cadro clínico, distínguense 4 formas de hemocromatosis hereditaria:
- I - tipo clásico autosómico recesivo, asociado ao HFE (máis do 95% dos casos)
- II - tipo xuvenil
- III - tipo hereditario asociado ao HFE (mutacións no receptor da transferrina tipo 2)
- IV - tipo dominante autosómico.
A hemocromatosis secundaria (hemosiderose xeneralizada) desenvólvese como consecuencia da insuficiencia adquirida dos sistemas enzimáticos implicados no metabolismo do ferro e adoita asociarse a outras enfermidades, en conexión coas que se distinguen as súas seguintes variantes: post-transfusión, nutricional, metabólica, mixta e neonatal.
No curso clínico, a hemocromatosis pasa por tres etapas: I - sen sobrecarga de ferro, II - con sobrecarga de ferro, pero sen síntomas clínicos, III - co desenvolvemento de manifestacións clínicas.

Causas da hemocromatosis
A hemocromatosis hereditaria primaria é un trastorno de transmisión autosómica recesiva. Baséase en mutacións do xene HFE localizado no brazo curto do 6º cromosoma. Un defecto no xen HFE leva a que as células do duodeno 12 mediadas pola transferrina mediadas por ferro polas células do duodeno resulten na formación dun falso sinal sobre a deficiencia de ferro no corpo. Á súa vez, isto contribúe a unha maior síntese da proteína de unión ao ferro DCT-1 por enterocitos e unha maior absorción de ferro no intestino (coa inxestión normal de oligoelementos dos alimentos). No futuro, prodúcese unha excesiva deposición do pigmento hemosiderina que contén ferro en moitos órganos internos, a morte dos seus elementos funcionalmente activos co desenvolvemento de procesos escleróticos. Con hemochromatosis, acumulan anualmente 0,5-1,0 g de ferro no corpo humano e as manifestacións da enfermidade maniféstanse cando se alcanza o nivel total de ferro de 20 g (ás veces 40-50 g ou máis).
A hemocromatosis secundaria desenvólvese como consecuencia dunha inxesta excesiva de exóxeno de ferro no corpo. Esta afección pode producirse con frecuentes transfusións de sangue repetidas, inxestión incontrolada de preparados de ferro, talasemia, algúns tipos de anemia, porfiria cutánea, cirrosis alcohólica do fígado, hepatite B e C crónicas, neoplasias malignas, seguindo unha dieta baixa en proteínas.
Síntomas de hemocromatosis
A manifestación clínica da hemocromatosis hereditaria prodúcese na idade adulta, cando o contido de ferro total no corpo alcanza valores críticos (20-40 g). Dependendo das síndromes predominantes, distínguense as formas hepatopáticas (hemocromatosis hepática), cardiopática (hemocromatosis cardíaca), as formas endocrinolóxicas da enfermidade.
A enfermidade desenvólvese gradualmente, na etapa inicial predominan queixas non específicas sobre aumento da fatiga, debilidade, perda de peso, diminución da libido. Nesta fase, os pacientes poden estar perturbados pola dor no hipocondrio dereito, pel seca, artralxia por condrocalcinose de grandes articulacións. Na etapa expandida da hemocromatosis, fórmase un complexo clásico de síntomas, representado pola pigmentación da pel (pel bronce), cirrosis, diabetes mellitus, cardiomiopatía, hipogonadismo.
Normalmente, o primeiro signo de hemocromatosis é a aparición dunha cor específica da pel e das mucosas, expresada principalmente na cara, pescozo, extremidades superiores, nas axilas e os xenitais externos e as cicatrices. A intensidade da pigmentación depende da duración do curso da enfermidade e varía desde o gris pálido (fumado) ata o marrón bronce. Característica é a perda de cabelo na cabeza e no tronco, deformación cóncava (en forma de culler) das uñas. As artropatías das articulacións metacarpofalangeas, ás veces do xeonllo, da cadeira e do cóbado notan co desenvolvemento posterior da súa rixidez.
En case todos os pacientes, detéctase un aumento no fígado, a esplenomegalia, a cirrosis do fígado. A disfunción do páncreas exprésase no desenvolvemento da diabetes mellitus dependente da insulina. Como resultado de danos na glándula hipofisaria durante a hemocromatosis, a función sexual padece: nos homes, desenvólvense atrofia testicular, impotencia, ginecomastia, nas mulleres - amenorrea e infertilidade. A hemocromatosis cardíaca caracterízase por cardiomiopatía e as súas complicacións - arritmia, insuficiencia cardíaca crónica, infarto de miocardio.
Na etapa terminal da hemocromatosis, desenvólvese hipertensión portal, ascite, cachéxia. Por regra xeral, a morte de pacientes prodúcese como sangrado por varices do esófago, insuficiencia hepática, insuficiencia cardíaca aguda, coma diabético, peritonite aséptica, sepsis. A hemocromatosis aumenta significativamente o risco de desenvolver cancro de fígado (carcinoma hepatocelular).
Diagnóstico de hemocromatosis
Dependendo dos síntomas prevalentes, os pacientes con hemocromatosis poden solicitar axuda de varios especialistas: gastroenterólogo, cardiólogo, endocrinólogo, xinecólogo, urólogo, reumatólogo e dermatólogo. Mentres tanto, o diagnóstico da enfermidade é o mesmo para varias variantes clínicas de hemocromatosis. Despois de avaliar os signos clínicos, os pacientes teñen asignados un conxunto de estudos de laboratorio e instrumentais para comprobar a validez do diagnóstico.
Os criterios de laboratorio para a hemocromatosis son un aumento significativo do nivel de ferro, ferritina e transferrina no soro sanguíneo, un aumento da excreción de ferro na urina e unha diminución da capacidade total de unión ao ferro do soro sanguíneo. O diagnóstico confírmase mediante biopsia de punción do fígado ou da pel, nas mostras das que se detecta a deposición de hemosiderina. A natureza hereditaria da hemocromatosis establécese como resultado dos diagnósticos xenéticos moleculares.
Para avaliar a gravidade dos danos nos órganos internos e o prognóstico da enfermidade, estanse estudando probas de fígado, niveis de glucosa en sangue e urina, hemoglobina glicosilada ... O diagnóstico de hemocromatosis en laboratorio compleméntase con estudos instrumentais: radiografía conxunta, ECG, ecocardiografía, ecografía da cavidade abdominal, resonancia magnética do fígado, etc.
Tratamento da hemocromatosis
O obxectivo principal da terapia é eliminar o exceso de ferro do corpo e evitar o desenvolvemento de complicacións. Os pacientes con hemocromatosis reciben unha dieta que restrinxa alimentos ricos en ferro (mazás, carne, fígado, trigo mouro, espinaca, etc.), hidratos de carbono facilmente dixeribles. Está prohibido tomar multivitaminas, ácido ascórbico, suplementos dietéticos que conteñan ferro, alcol. Para eliminar o exceso de ferro do corpo, recorren á sangría baixo o control da hemoglobina, o hematocrito e a ferritina. Para este propósito, pódense empregar métodos de hemocorrección extracorpórea: plasmafereis, hemosorción, citofereza.
A terapia farmacoxenética da hemocromatosis baséase na administración intramuscular ou intravenosa de ións Fe3 + que unen a deferoxamina a un paciente. Ao mesmo tempo, realízase un tratamento sintomático de cirrosis do fígado, insuficiencia cardíaca, diabetes mellitus e hipogonadismo. Con artropatía grave, determínanse indicacións para a artroplastia (endoprotétesis das articulacións afectadas). En pacientes con cirrosis, estase a tratar a cuestión do transplante de fígado.
Predición e prevención da hemocromatosis
A pesar do curso progresivo da enfermidade, a terapia oportuna pode estender a vida dos pacientes con hemocromatosis durante varias décadas. A falta de tratamento, a esperanza de vida media dos pacientes despois do diagnóstico da patoloxía non supera os 4-5 anos. A presenza de complicacións da hemocromatosis (principalmente cirrosis hepática e insuficiencia cardíaca conxestiva) é un signo prognosticamente desfavorable.
A hemocromatosis hereditaria, a prevención descríbese na detección familiar, na detección precoz e no tratamento da enfermidade. A nutrición racional, o control da administración e administración de preparacións de ferro, transfusións de sangue, a negativa a beber alcohol e o seguimento dos pacientes con enfermidades do fígado e do sistema sanguíneo permiten evitar o desenvolvemento de hemocromatosis secundaria.